
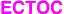 Synthetic Papers and Posters
Synthetic Papers and Posters
M. David Weingarten and Albert Padwa*
Department of Chemistry, Emory University, Atlanta, Georgia 30322


Albert padwa
The construction of ring systems by intramolecular addition of an anionic center to a carbon-carbon pi-bond has attracted considerable attention in recent years [1-7]. Various organometallic reagents have been used resulting in the formation of cycloalkenyl containing products. The synthetic utility of these anionic cyclizations is further enhanced by the ease with which the organometallic product may be functionalized by reaction with various electrophiles. The demonstration by Dunitz and Burgi [8] of favored trajectories for the approach of one reactant molecule toward another led to the formulation of rules governing the ease of intramolecular ring closure reactions [9]. Cyclization of the 5-hexenyl anion is predicted to occur by way of a 5-exo-trig closure since this pathway permits the optimum trajectory by the nucleophile of 109 deg to the double bond in the plane of its pi-orbitals. For cyclizations involving nucleophilic attack at triple bonds, the situation remains less clear-cut than for the analogous ring closures in tetrahedral or trigonal systems. The original rules [9] postulated an acute approach angle of about 60 deg in digonal systems and stated that endo-dig ring closures are generally preferred for the formation of five and six-membered rings. Since that time, there have been several theoretical studies which indicate that the favored path of approach of a nucleophile to a triple bond is at an obtuse angle of 120-127 degrees [10-14]. In the case of electronically unbiased acetylenes, exo-dig cyclizations are favored [15]. Thus, Bailey and coworkers have repeatedly shown that 5-alkyn-1-yl lithiums prefer to undergo anionic cyclization via a highly regiospecific 5-exo-dig process involving stereoselective syn-addition to the triple bond [4]. A wide variety of compounds of type 1
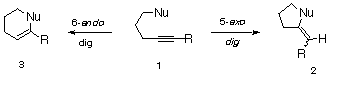

undergo exclusive 5-exo-dig cyclization to give cyclopentenyl (2) rather than cyclohexenyl (3) products [16-19]. Since questions of angle of approach of an internal nucleophile to the triple bond as well as the degree of involvement of the carbonyl group along the reaction pathway are still of considerable interest, we decided to carry out an exploratory study on the base-induced cyclizations of o-ethynylaryl benzylic alcohols of type 4-9. As far as we know, this reaction has not yet been investigated in any detail, in spite of the ability of unactivated alkynes to undergo nucleophilic additions with alkoxide ions. Our results are relevant to the understanding of cyclizations involving nucleophilic attack at triple bonds.
Under basic conditions, the hydroxyl functionality of alkynes 4-9 underwent smooth cyclization with the unactivated acetylenic group. Much to our surprise, the mode of cyclization seemed to be greatly influenced by the nature of the substituent in the ortho-position of the aromatic ring. For example, treatment of alkynyl alcohol 5 (R=o-CO2Me) with NaH in THF produced benzopyran 11 in >90% yield. The formation of 11 corresponds to a rare example of a 6-endo-dig cyclization. On the other hand, treatment of the related alkynyl alcohols 4 (R=H) and 6 (R=o-OMe) under the same conditions produced only the 5-exo-dig products 10 and 12 in quantitative yield. Since it appeared that the 6-endo cyclization of these benzylic alcohols was dependent on the presence of an electron-withdrawing group in the ortho-position, we prepared the corresponding alkynyl alcohols 7 (R=o-CHO), 8 (R=o-NO2), and 9 (R=p-CO2Me). The 6-endo-dig cyclization product 13 was exclusively formed with 7, but only the 5-exo-digprocess occurred with 8 and 9.
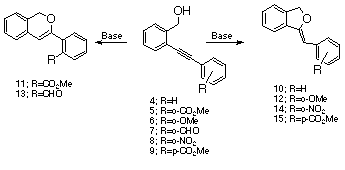

The fact that 6-endo cyclization mode only occurs when a carbonyl group is present in the ortho-position of the aromatic ring prompted us to examine the reaction in greater detail. Further study showed that the product distribution was found to be markedly dependent upon the reaction conditions. Careful monitoring of the crude mixture of 5 and 7 by NMR spectroscopy indicated that the reaction proceeded by initial formation of the 5-exo-dig product followed by subsequent rearrangement to the 6-endo product. Thus, these apparent 6-endo cyclizations are actually the consequence of a 5-exo cyclization followed by a rapid acid-catalyzed rearrangement. Although we were unable to isolate benzofurans 16 and 17 due to their facile conversion to benzopyrans 11 and 13, their structures were confirmed by ozonolysis of the crude reaction mixture to phthalide and the corresponding aryl aldehydes.
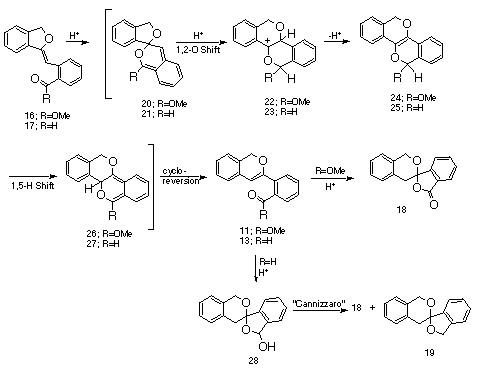
A plausible mechanism for the acid-catalyzed rearrangment of 16 to 11 is outlined below. The first step involves an initial protonation of benzofuran 16 followed by intramolecular cyclization of the ortho-carbonyl group to form spiro ketal 20 which then proceeds to benzopyran 11 via a series of reactions. Ring expansion by a 1,2-O shift results in the formation of cation 22 which undergoes proton loss to produce 24. This intermediate proceeds to the final product by a rapid 1,5-sigmatropic hydrogen shift and a subsequent cycloreversion. Further reaction of 11 with acid results in the formation of spiro lactone18. Benzofuran 17 undergoes a related sequence ultimately affording lactol 28 which is converted to lactone 18 and spiro ketal 19 via an acid-catalyzed Cannizzaro reaction [20].
Further studies on the mechanistic details and synthetic potential of these cyclizations are in progress.
Acknowledgment: M.D.W is the recipient of a Graduate Fellowship from the Organic Chemistry Division of the American Chemical Society (1994-1995) sponsored by Proctor & Gamble, Co. We wish to thank the National Science Foundation for generous support of this research.
References and Notes:
1. Kossa, W. C.; Rees, T. C.; Richey, H. G. Tetrahedron Lett. 1971, 3455-8.
2. St. Denis, J.; Oliver, J. P.; Dolzine, T. W.; Smart, J. B. J. Organomet. Chem. 1974, 71, 315-23. Dolzine, T.; Oliver, J. P. J. Organomet. Chem. 1974, 78, 165-76.
3. Crandall, J. K.; Battioni, P.; Wehlacz, J. T.; Bindra, R. J. Am. Chem. Soc. 1975, 97, 7171-2.
4. Bailey, W. F.; Khanolkar A. D.; Gavaskar, K. V. J. Am. Chem. Soc. 1992, 114, 8053-60.
5. Cooke, Jr., M. P. J. Org. Chem. 1992, 57, 1495-1503.
6. Chamberlin, A. R.; Bloom, S. H.; Cervini, L. A.; Fotsch, C. H. J. Am. Chem. Soc. 1988, 110, 4788-95.
7. Paquette, L. A.; Gilday, J. P.; Maynard, G. D. J. Org. Chem. 1989, 54, 5044-53.
8. Bürgi, H. B.; Dunitz, J. D. Acc. Chem. Res. 1983, 16, 153-61.
9. Baldwin, J. E. J. Chem. Soc., Chem. Commun. 1976, 734-6. Baldwin, J. E.; Cutting, J.; Dupont, W.; Kruse, L.; Silberman, L.; Thomas, R. C. J. Chem. Soc., Chem. Commun. 1976, 736-8. Baldwin, J. E.Further Perspectives in Organic Chemistry; A Ciba Foundation Symposium; Elsevier: Amsterdam, 1978, p. 85.
10. Perkins, M. J.; Wong, P. C.; Barrett, J.; Shalival, G. J. Org. Chem. 1981, 46, 2196-9.
11. Ersenstein, O.; Procter, G.; Dunitz, J. D. Helv. Chim. Acta. 1978, 61, 2538-41.
12. Dykstra, C. E.; Arduengo, A. J.; Fukunaga, F. T. J. Am. Chem. Soc. 1978, 100, 6007-12.
13. Strozier, R. W.; Caramella, P.; Houk, K. N. J. Am. Chem. Soc. 1979, 101, 1340-2. Houk, K. N.; Strozier, R. W.; Rozeboom, M. D.; Nagaze, S. J. Am. Chem. Soc. 1982, 104, 323-5.
14. Elliott, R. J.; Richards, W. G. J. Mol. Struct. Theochem. 1982, 87, 247.
15. Mellor, M.; Santos, A.; Swrell, E. G.; Sutherland, J. K. J. Chem. Soc., Chem. Commun. 1978, 528-30.
16. Evans, C. M.; Kirby, A. J. J. Chem. Soc., Perkin Trans. 2 1984, 1269-75.
17. Trost, B. M.; Runge, T. A. J. Am. Chem. Soc. 1981, 103, 7559-72.
18. Garcia, H.; Iborro, S.; Primo, J.; Miranda, M. A. J. Org. Chem. 1986, 51, 4432-6.
19. Brennan, C. M.; Johnson, C. D.; McDonnell, P. D. J. Chem. Soc., Perkin Trans. II 1989, 957-61.
20. Rieche, A.; Schmitz, E. Chem. Ber. 1957, 90, 531-4.

Experimental
Preparation of Methyl 2-[2-(Hydroxymethyl)phenylethynyl]benzoate (5). To a deareated solution containing 0.82 g (3.5 mmol) of 2-iodobenzyl alcohol and 0.60 g (4.16 mmol) of methyl 2-ethynylbenzoate in 50 mL of triethylamine was added 20 mg of bis-triphenylphosphine palladium (II) chloride and 20 mg of CuI under an argon atmosphere. The reaction mixture was heated at reflux for 12 h. After cooling, the mixture was filtered and concentrated under reduced pressure. Chromatography of the resulting brown oil on silica gel gave 0.75 g (80%) of methyl 2-[2-(hydroxymethyl)phenylethynyl]benzoate (5); IR (CH2Cl2) 1717, 1294, 1255, 1076, and 756 cm-1; 1H-NMR (CDCl3, 300 MHz) ppm 3.90 (s, 3H), 4.11 (t, 1H, J=6.3 Hz), 4.82 (d, 2H, J=6.3 Hz), 7.23 -7.40 (m, 4H), 7.49 (t, 1H, J=7.5 Hz), 7.56 (d, 1H, J= 7.0 Hz), 7.64 (d, 1H, J=7.6 Hz), and 7.98 (d, 1H, J=7.9 Hz); 13C-NMR (CDCl3, 75 MHz) ppm 52.4, 63.9, 92.3, 92.5, 121.8, 123.8, 127.3, 127.9, 128.1, 128.8, 130.4, 130.6, 131.9, 132.5, 134.2, 143.5, and 166.4; HRMS Calcd for C17H14O3: 266.0943. Found: 266.0922.
Reaction of Methyl 2-[2-(Hydroxymethyl)phenylethynyl]benzoate (5) with Sodium Hydride. To a solution of 40 mg of NaH (60%) in 10 mL of anhydrous THF was added a solution of 200 mg (0.75 mmol) of 5 in 10 mL of anhydrous THF under N2. After stirring for 6 h at rt, the reaction was concentrated on the rotary evaporator to give 1-(2-carbomethoxy-benzylidene) -1,3-dihydro-isobenzofuran (16) as an extremely labile compound whose 1H-NMR contains singlets at 3.84 (3H), 5.33 (2H) and 6.58 (1H). The structural assignment of 16 was verified by ozonolysis to give phthalide and methyl 2-formylbenzoate which matched authentic samples in all regards.
Benzofuran 16 was treated with a solution of saturated ammonium chloride and immediately extracted with CH2Cl2. The combined CH2Cl2 extracts were washed with water and brine and then dried over magnesium sulfate. Evaporation of the solvent under reduced pressure afforded 120 mg (60%) of methyl 2-(1H-2-benzo-pyran-3-yl)benzoate (11); mp 72-73 oC; IR (CH2Cl2) 1716, 1648, 1485, 1258, and 1037 cm-1; 1H-NMR (CDCl3, 300 MHz) ppm 3.92 (s, 3H), 5.47 (s, 2H), 7.06 (s, 1H), 7.16 (t, 1H, J=7.6 Hz), 7.25-7.40 (m, 3H), 7.48 (t, 1H, J=7.4 Hz), 7.67 (m, 1H), 7.91 (d, 1H, J=8.0 Hz), and 8.39 (d, 1H, J=8.0 Hz); 13C-NMR (CDCl3) ppm 51.8, 74.9, 92.9, 120.5, 121.0, 124.6, 127.0, 128.0, 129.0, 129.3, 130.5, 131.6, 134.8, 137.1, 139.2, 157.5, and 168.3; Anal Calcd for C17H14O3: C, 76.66; H, 5.30. Found: C, 76.53; H, 5.22.
Reaction of Methyl 2-(1H-2-benzo-pyran-3-yl)benzoate (11) with Acid. Benzopyran 11 was further treated with a saturated ammonium chloride solution and stirred at rt for 10 min. The combined CH2Cl2 extracts were washed with brine, dried over magnesium sulfate, and concentrated under reduced pressure. Chromotography of the residue on silica gel afforded 3'-oxo-spiro[3H-1,4-dihydro-2-benzopyran-3,1'(3'H)-isobenzofuran] (18); mp 209-210 oC; IR (neat) 2030, 2868, 1700, and 1603 cm-1; 1H-NMR (300 MHz, CDCl3) 3.58 (AB, 2H, J=16.2 Hz), 5.16 (AB, 2H, J=12.9 Hz), 7.30-7.65 (m, 7H), and 8.16 (d, 1H, J=7.8 Hz); 13C-NMR (75 MHz, CDCl3) 36.9, 72.7, 112.8, 121.4, 122.1, 124.9, 127.8, 18, 128.1, 130.1, 130.2, 134, 136.5, 137.7, 139.9, and 164.2; Anal. Calcd. for C16H12O3: C, 76.18; H, 4.79. Found: C, 76.08; H, 4.84.
 DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
http://newdrugapprovals.org/
DRUG APPROVALS BY DR ANTHONY MELVIN CRASTO
No comments:
Post a Comment