
1 RAGAGLITAZAR
2 TROGLITAZONE
3 MITOGLITAZONE
4 PIOGLITAZONE
5 NAVEGLITAZAR
6 SAROGLITAZAR
7 CIGLITAZONE
8
9
10
2 MURA
3 INDE
Indeglitazar


1
Ragaglitazar ........Dr. Reddy's Research Foundation
1
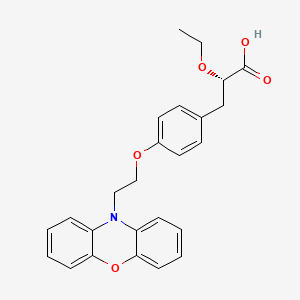


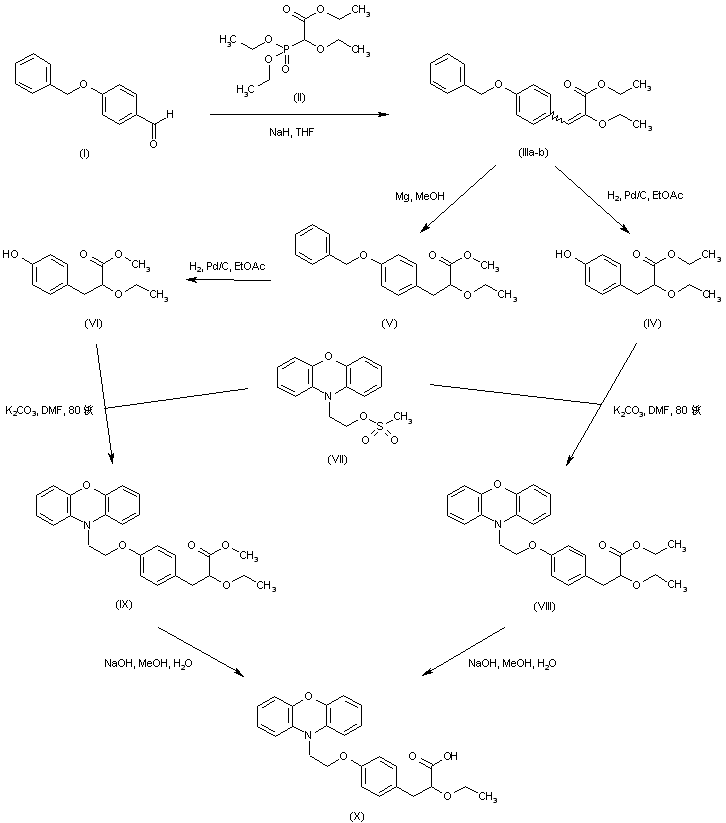

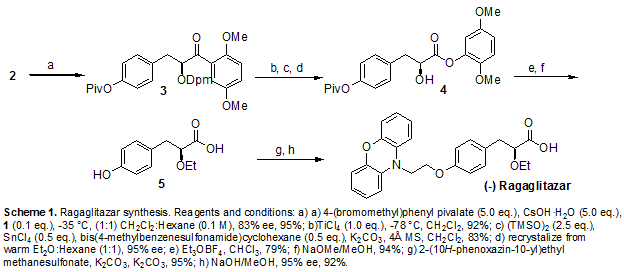






| PATENT | SUBMITTED | GRANTED |
|---|---|---|
| Benzamides as ppar modulators [US2006160894] | 2006-07-20 | |
| Novel tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US2002077320] | 2002-06-20 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US7119198] | 2006-07-06 | 2006-10-10 |
| Tricyclic compounds and their use in medicine: process for their preparation and pharmaceutical compositions containing them [US6440961] | 2002-08-27 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6548666] | 2003-04-15 | |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6608194] | 2003-08-19 | |
| CRYSTALLINE R- GUANIDINES, ARGININE OR (L) -ARGININE (2S) -2- ETHOXY -3-{4- [2-(10H -PHENOXAZIN -10-YL)ETHOXY]PHENYL}PROPANOATE [WO0063189] | 2000-10-26 | |
| Pharmaceutically acceptable salts of phenoxazine and phenothiazine compounds [US6897199] | 2002-11-14 | 2005-05-24 |
| Tricyclic compounds and their use in medicine process for their preparation and pharmaceutical compositions containing them [US6939988] | 2005-09-06 |
wo/2014/181362 a process for the preparation of 3 … – WIPO
patentscope.wipo.int/search/en/WO2014181362
Nov 13, 2014 - (WO2014181362) A PROCESS FOR THE PREPARATION OF 3-ARYL-2-HYDROXY PROPANOIC ACID COMPOUNDS …
- Dolling, U. H.; Davis, P.; Grabowski, E. J. J. Efficient Catalytic Asymmetric Alkylations. 1. Enantioselective Synthesis of (+)-Indacrinone via Chiral Phase-Transfer Catalysis. J. Am. Chem. Soc. 1984, 106, 446–447.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C. Phase-Transfer Catalyzed Asymmetric Glycolate Alkylation. Org. Lett. 2004, 6, 2289–2292.
- Andrus, M. B.; Hicken, E. J.; Stephens, J. C.; Bedke, D. K. Asymmetric Phase-Transfer Catalyzed Glycolate Alkylation, Investigation of the Scope, and Application to the Synthesis of (-)-Ragaglitazar. J. Org. Chem. 2005, ASAP.
- Henke, B. R. Peroxisome Proliferator-Activated Receptor Dual Agonists for the Treatment of Type 2 Diabetes. J. Med. Chem. 2004, 47, 4118–4127.
- Wilson, T. M.; Brown, P. J.; Sternbach, D. D.; Henke, B. R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000, 46, 1306–1317.
- Uchida, R.; Shiomi, K.; Inokoshi, J.; Masuma, R.; Kawakubo, T.; Tanaka, H.; Iwai, Y.; Omura, A. Kurasoins A and B, New Protein Farnesyltrasferase Inhibitors Produced by Paecilomyces sp. FO-3684. J. Antibio. 1996, 49, 932–934.

- CI 991
- CS 045
- Depotox
- GR 92132X
- Noscal
- Rezulin
- Romglizone
- Troglitazone
 Type-II diabetes mellitus (DM) is characterized by insulin resistance, glucose intolerance, increased hepatic glucose production, and decreased pancreatic insulin secretion. In the past, the drug classes used for type-II DM have targeted the last three of these abnormalities. Sulfonylurea agents bind to ATP-dependent potassium efflux channels to stimulate pancreatic insulin secretion at b-islet cells. The biguanides decrease hepatic glucose production, and thea-glucosidase inhibitors delay carbohydrate digestion to improve glucose tolerance. Until the recent advent of the thiazolidinedione drugs (ciglitazone was first synthesized in 1982), there was no therapy specifically targeting insulin resistance. Drugs of this class all share a common thiazolidine-2-4-dione structure. Marketed drugs of this class include pioglitazone, rosiglitazone, and troglitazone [Figure 1] – the first to reach the market.
Type-II diabetes mellitus (DM) is characterized by insulin resistance, glucose intolerance, increased hepatic glucose production, and decreased pancreatic insulin secretion. In the past, the drug classes used for type-II DM have targeted the last three of these abnormalities. Sulfonylurea agents bind to ATP-dependent potassium efflux channels to stimulate pancreatic insulin secretion at b-islet cells. The biguanides decrease hepatic glucose production, and thea-glucosidase inhibitors delay carbohydrate digestion to improve glucose tolerance. Until the recent advent of the thiazolidinedione drugs (ciglitazone was first synthesized in 1982), there was no therapy specifically targeting insulin resistance. Drugs of this class all share a common thiazolidine-2-4-dione structure. Marketed drugs of this class include pioglitazone, rosiglitazone, and troglitazone [Figure 1] – the first to reach the market.Approval history
Lawsuits
Mode of action


| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (RS)-5-(4-[(6-hydroxy-2,5,7,8-tetramethylchroman-2-yl)methoxy]benzyl)thiazolidine-2,4-dione | |
| CLINICAL DATA | |
| LEGAL STATUS |
?
|
| PHARMACOKINETIC DATA | |
| HALF-LIFE | 16-34 hours |
| IDENTIFIERS | |
| CAS NUMBER | 97322-87-7 |
| ATC CODE | A10BG01 |
| PUBCHEM | CID 5591 |
| IUPHAR LIGAND | 2693 |
| DRUGBANK | DB00197 |
| CHEMSPIDER | 5389 |
| UNII | I66ZZ0ZN0E |
| KEGG | D00395 |
| CHEBI | CHEBI:9753 |
| CHEMBL | CHEMBL408 |
| CHEMICAL DATA | |
| FORMULA | C24H27NO5S |
| MOL. MASS | 441.541 g/mol |






References
- Fisher, Lawrence (4 November 1997). “Adverse Diabetes Drug News Sends Warner-Lambert Down”. The New York Times. Retrieved 12 December 2012.
- Retired Drugs: Failed Blockbusters, Homicidal Tampering, Fatal Oversights, wired.com
- Cohen, J. S. (2006). “Risks of troglitazone apparent before approval in USA”.Diabetologia 49 (6): 1454–5. doi:10.1007/s00125-006-0245-0. PMID 16601971.
- Henry RR (September 1996). “Effects of troglitazone on insulin sensitivity”. Diabet. Med.13 (9 Suppl 6): S148–50.PMID 8894499.
- Keen H (November 1994). “Insulin resistance and the prevention of diabetes mellitus”. N. Engl. J. Med. 331 (18): 1226–7.doi:10.1056/NEJM199411033311812. PMID 7935664.
- Willman, David (20 December 2000). “NEW FDA: Rezulin Fast-Track Approval and a Slow Withdrawal”. The Los Angeles Times. Retrieved 12 December 2012.
- Willman, David (4 June 2000). “The Rise and Fall of the Killer Drug Rezulin”. The Los Angeles Times. Retrieved 12 December 2012.
- “Report: FDA Removes Medical Officer”.
- Avorn, J (2005). Powerful medicines. New York: Vintage books.
- Gøtzsche, Peter (2013). Deadly medicines and organised crime : how big pharma has corrupted healthcare. London [u.a.]: Radcliffe Publ. p. 185. ISBN 9781846198847.
- Leary, Warren (31 January 1997). “New Class of Diabetes Drug Is Approved”. The New York Times. Retrieved 12 December 2012.
- Sinclair, Neil (31 July 1997). “Glaxo Wellcome gets approval for Romozin”. ICIS News. Retrieved 12 December 2012.
- “www.accessdata.fda.gov”.
- British Broadcasting Corporation (1 December 1997). “Diabetes drug withdrawn from sale”. BBC. Retrieved 12 December 2012.
- Fisher, Lawrence (17 January 1998). “Drug Makers at Threshold of a New Therapy; With a Dose of Biotechnology, Big Change Is Ahead in the Treatment of Diabetes”. The New York Times. Retrieved 12 December 2012.
- Diabetes Prevention Research Group (April 1999). “Design and methods for a clinical trial in the prevention of type 2 diabetes”.Diabetes Care 22 (4): 623–634.doi:10.2337/diacare.22.4.623. Retrieved 12 December 2012.
- Diabetes Prevention Program Research Group (7 February 2002). “Reduction in the Incidence of Type 2 Diabetes with Lifestyle Intervention or Metformin”. The New England Journal of Medicine 346 (6): 393–403.doi:10.1056/NEJMoa012512.PMC 1370926. PMID 11832527. Retrieved 12 December 2012.
- Gale, Edwin (January 2006). “Troglitazone: the lesson that nobody learned?”.Diabetologia 49 (1): 1–6. doi:10.1007/s00125-005-0074-6.
- Willman, David (16 August 2000). “FDA’s Approval and Delay in Withdrawing Rezulin Probed”. The Los Angeles Times. Retrieved 12 December 2012.
- Willman, David (10 March 2000). “Fears Grow Over Delay in Removing Rezulin”. The Los Angeles Times. Retrieved 12 December 2012.
- U.S. Food and Drug Administration. “2000 Safety Alerts for Human Medical Products”. U.S. Food and Drug Administration. Retrieved 12 December 2012.
- Willman, David (March 17, 2000). “Physician Who Opposes Rezulin Is Threatened by FDA With Dismissal”. Los Angeles Times.
- Pfizer. “Pfizer Annual Report 2001″. Pfizer. Retrieved 12 December 2012.
- Feeley, Jef (March 31, 2009). “Pfizer Ends Rezulin Cases With $205 Million to Spare”.Bloomberg. Retrieved 6 April 2014.
- Aljada A, Garg R, Ghanim H, et al. (2001). “Nuclear factor-kappaB suppressive and inhibitor-kappaB stimulatory effects of troglitazone in obese patients with type 2 diabetes: evidence of an antiinflammatory action?”. J. Clin. Endocrinol. Metab. 86 (7): 3250–6.doi:10.1210/jc.86.7.3250. PMID 11443197.
External links
- Diabetes Monitor article on troglit
| US4316849 * | 11 Jul 1980 | 23 Feb 1982 | Blasinachim S.P.A. | Process for preparing a crystalline polymorphous type of chenodeoxycholic acid |
| US4572912 * | 28 Aug 1984 | 25 Feb 1986 | Sankyo Company Limited | Treatment of hyperlipemia and hyperglycemia |
| US5248699 * | 13 Aug 1992 | 28 Sep 1993 | Pfizer Inc. | Hydrochloride salt, antidepressant, anorectic |
| US5319097 * | 11 Dec 1991 | 7 Jun 1994 | Imperial Chemical Industries Plc | Pharmaceutical agents |
| AU3255984A* | Title not available | |||
| EP0014590A1* | 7 Feb 1980 | 20 Aug 1980 | Eli Lilly And Company | Crystalline forms of N-2-(6-methoxy)benzothiazolyl-N’-phenyl urea and process for their preparation |
| EP0022527A1* | 4 Jul 1980 | 21 Jan 1981 | BLASCHIM S.p.A. | Process for preparing a solvent-free crystalline polymorphous form of chenodeoxycholic acid |
| EP0490648A1* | 11 Dec 1991 | 17 Jun 1992 | Zeneca Limited | Pharmaceutical agents |
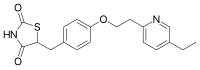
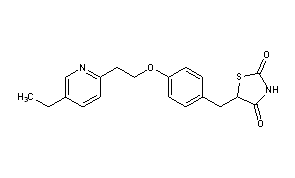
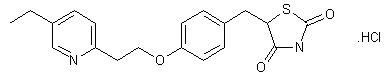
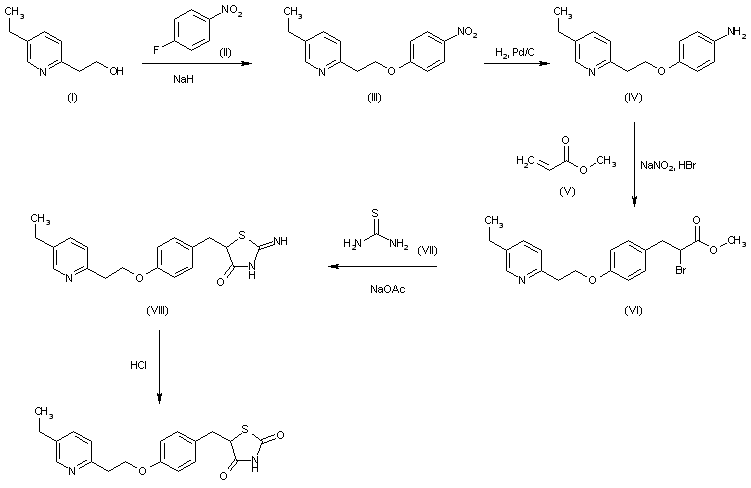
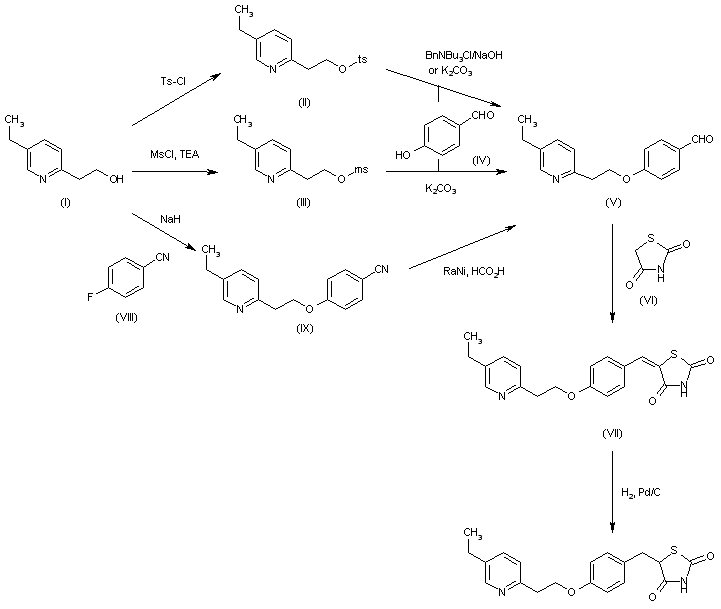
MITOGLITAZONE
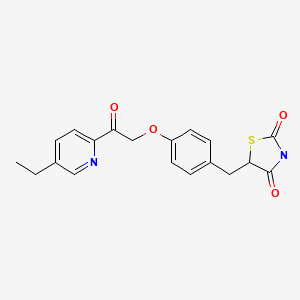 MITOGLITAZONE MSDC-0160; CAY 10415; 146062-49-9 5-[4-[2-(5-Ethylpyridin-2-yl)-2-oxoethoxy]benzyl]thiazolidine-2,4-dione 5-[[4-[2-(5-ethylpyridin-2-yl)-2-oxoethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione 5-(4-(2-(5-cthylpyridin-2-yl)- 2-oxoethoxy)benzyl)-1,3 -thiazolidiiie-2,4-dione Pfizer, INNOVATOR phase 2 MSD-9 MSDC-0160 PNU-91325 U-91325 BACKGROUND Thiazolidinedione analogs either prevent or ameliorate an insulin resistance state, which occurs genetically or is induced by dietary means. 5-(4-(2-(5-Ethylpyridin-2-yl)-2- oxoethoxy)benzyl)-l,3-thiazolidine-2,4-dione of Formula (I) (Mitoglitazonc) is an antidiabetic thiazolidinedione being evaluated for the treatment of non-insulin-dependent diabetes mellitus. Non-insulin-dependent diabetes mellitus (NIDDM) is a metabolic disease characterized by a reduction in the response of the peripheral target tissue to insulin and the inability of pancreatic insulin reserves to overcome the reduced response. Improvement of insulin sensitivity of the target tissue not only reduces the consequences of the disease but actually aids in the prevention of NIDDM. The synthesis of Mitoglitazone has been reported (J. Med. Chem. , 1996, 39, 5053- 5063; Org. Pro. Res. & Dev. (OPRD), 2002, 6, 721-728 and U.S. PaL No. 5,441,971). .................................. http://www.google.com/patents/WO1992018501A1?cl=en
MITOGLITAZONE MSDC-0160; CAY 10415; 146062-49-9 5-[4-[2-(5-Ethylpyridin-2-yl)-2-oxoethoxy]benzyl]thiazolidine-2,4-dione 5-[[4-[2-(5-ethylpyridin-2-yl)-2-oxoethoxy]phenyl]methyl]-1,3-thiazolidine-2,4-dione 5-(4-(2-(5-cthylpyridin-2-yl)- 2-oxoethoxy)benzyl)-1,3 -thiazolidiiie-2,4-dione Pfizer, INNOVATOR phase 2 MSD-9 MSDC-0160 PNU-91325 U-91325 BACKGROUND Thiazolidinedione analogs either prevent or ameliorate an insulin resistance state, which occurs genetically or is induced by dietary means. 5-(4-(2-(5-Ethylpyridin-2-yl)-2- oxoethoxy)benzyl)-l,3-thiazolidine-2,4-dione of Formula (I) (Mitoglitazonc) is an antidiabetic thiazolidinedione being evaluated for the treatment of non-insulin-dependent diabetes mellitus. Non-insulin-dependent diabetes mellitus (NIDDM) is a metabolic disease characterized by a reduction in the response of the peripheral target tissue to insulin and the inability of pancreatic insulin reserves to overcome the reduced response. Improvement of insulin sensitivity of the target tissue not only reduces the consequences of the disease but actually aids in the prevention of NIDDM. The synthesis of Mitoglitazone has been reported (J. Med. Chem. , 1996, 39, 5053- 5063; Org. Pro. Res. & Dev. (OPRD), 2002, 6, 721-728 and U.S. PaL No. 5,441,971). .................................. http://www.google.com/patents/WO1992018501A1?cl=en  ..........................................
..........................................
 13C NMR PREDICTION
13C NMR PREDICTION  ....
....

Medical uses
| (RS)-5-(4-[2-(5-ethylpyridin-2-yl)ethoxy]benzyl)thiazolidine-2,4-dione | |
| CLINICAL DATA | |
|---|---|
| TRADE NAMES | Actos |
| AHFS/DRUGS.COM | monograph |
| MEDLINEPLUS | a699016 |
| LICENCE DATA | EMA:Link, US FDA:link |
| PREGNANCY CAT. |
|
| LEGAL STATUS | |
| ROUTES | oral |
| PHARMACOKINETIC DATA | |
| PROTEIN BINDING | >99% |
| METABOLISM | liver (CYP2C8) |
| HALF-LIFE | 3–7 hours |
| EXCRETION | in bile |
| IDENTIFIERS | |
| CAS NUMBER | 111025-46-8 |
| ATC CODE | A10BG03 |
| PUBCHEM | CID 4829 |
| IUPHAR LIGAND | 2694 |
| DRUGBANK | DB01132 |
| CHEMSPIDER | 4663 |
| UNII | X4OV71U42S |
| KEGG | D08378 |
| CHEBI | CHEBI:8228 |
| CHEMBL | CHEMBL595 |
| CHEMICAL DATA | |
| FORMULA | C19H20N2O3S |
| MOL. MASS | 356.44 g/mol |
| US PATENT NO | EXPIRY DATE | PATENT USE CODE |
|---|---|---|
| 5965584 | Jun 19, 2016 | U-753 |
| 6150383 | Jun 19, 2016 | U-418 |
| 6150384 | Jun 19, 2016 | U-419 |
| 6166042 | Jun 19, 2016 | U-414 |
| 6166043 | Jun 19, 2016 | U-415 |
| 6172090 | Jun 19, 2016 | U-416 |
| 6211205 | Jun 19, 2016 | U-410 |
| 6271243 | Jun 19, 2016 | U-411 |
| 6303640 | Aug 9, 2016 | U-425 |
| 6329404 | Jun 19, 2016 | U-753 |
| DMF16635 | STATUSA | TYPEII | SUMISSION6/11/2003 | BIOCON LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN BALGALORE, INDIA. |
| 16672 | A | II | 6/30/2003 | CIPLA LTD | PIOGLITAZONE HYDROCHLORIDE USP (ESUB) AS MANUFACTURED IN PUNE, MAHARASHTRA, INDIA |
| 16675 | A | II | 7/2/2003 | DR REDDYS LABORATORIES LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN ANDHRA PRADESH, INDIA. |
| 16682 | A | II | 7/3/2003 | RANBAXY LABORATORIES LTD | PIOGLITAZONE HYDROCHLORIDE AS MANUFACTURED IN PUNJAB, INDIA. |
| 16684 | A | II | 7/8/2003 | USV LIMITED | PIOGLITAZONE HYDROCHLORIDE (ESUB) AS MANUFACTURED IN MUMBAI, INDIA. |








![[1860-5397-9-265-i12]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i12.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i13]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i13.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i14]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i14.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i15]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i15.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i16]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i16.png?scale=2.0&max-width=1024&background=FFFFFF)
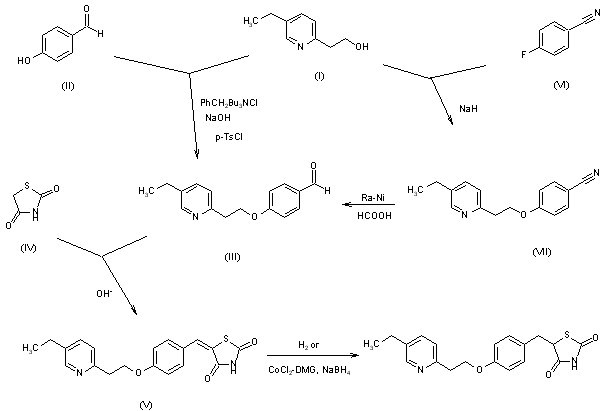
48 Halama, A.; Hejtmankova, L.; Lustig, P.; Richter, J.; Srsnova, L.; Jirman, J. Method for obtaining Pioglitazone as an antidiabetic agent. W.O. Patent WO02088120, Nov 7, 2002.



| US8067450 | Sep 15, 2008 | Nov 29, 2011 | Metabolic Solutions Development Company | Thiazolidinedione analogues for the treatment of metabolic diseases |
| US8304441 | Nov 16, 2011 | Nov 6, 2012 | Metabolic Solutions Development Company, Llc | Thiazolidinedione analogues for the treatment of metabolic diseases |
| WO2011017244A1* | Aug 2, 2010 | Feb 10, 2011 | Metabolic Solutions Development Company | Polymorphs of 5-(4-(2-(5-ethylpyridin-2-yl)-2-oxoethoxy)benzyl)-1,3-thiazolidine-2,4-dione (mitoglitazone) |
| WO2012153312A1 | May 11, 2012 | Nov 15, 2012 | Ranbaxy Laboratories Limited | Process for the purification of pioglitazone |
| WO2003053367A2 | Dec 20, 2002 | Jul 3, 2003 | Ben-Zion Dolitzky | Hydrogenation of precursors to thiazolidinedione antihyperglycemics |
| WO2003080056A2 | Mar 21, 2003 | Oct 2, 2003 | Ben-Zion Dolitzky | Fine particle size pioglitazone |
| WO2004007490A2 | Jul 15, 2003 | Jan 22, 2004 | Cadila Healthcare Ltd | A process to prepare pioglitazone via several intermediates. |
| WO2004024059A2 | Aug 29, 2003 | Mar 25, 2004 | Parag Narayan Gadkari | Improved process for preparation of thiazolidinedione derivatives |
| WO2004101560A1 | May 11, 2004 | Nov 25, 2004 | Frantisek Picha | Processes for making thiazolidinedione derivatives and compounds thereof |
| WO2004101561A1 | May 13, 2004 | Nov 25, 2004 | Frantisek Picha | Pioglitazone salts, such as pioglitazone sulfate, and pharmaceutical compositions and processes using the same |
| WO2005049610A1 | Oct 27, 2004 | Jun 2, 2005 | Sudhir Nambiar | Process for preparing thiazolidinediones |
| WO2005058827A1 | Dec 16, 2004 | Jun 30, 2005 | Janos Fischer | Process for the synthesis of pioglitazone hydrogen chloride |
| WO2006117654A1 | May 3, 2006 | Nov 9, 2006 | Ranbaxy Lab Ltd | Processes for the preparation of pioglitazone or salts thereof |
| WO2008075380A2 | Dec 20, 2007 | Jun 26, 2008 | Ind Swift Lab Ltd | Process for the preparation of thiazolidine derivatives |
| WO2008142706A2 | May 19, 2008 | Nov 27, 2008 | Yogendra Kumar Chauhan | Novel process for the synthesis of pioglitazone and its salts thereof |
| WO2009104200A1 | Apr 8, 2008 | Aug 27, 2009 | Biocon Limited | A method for preparation of thiazolidinedione derivatives |
| WO2009133576A1 | Apr 28, 2008 | Nov 5, 2009 | Erregierre S.P.A. | A process for the preparation of 4-[2-(5-ethyl-2-pyridyl)ethoxy]nitrobenzene and pioglitazone |
| US4687777 | Jan 17, 1986 | Aug 18, 1987 | Takeda Chemical Industries, Ltd. | Thiazolidinedione derivatives, useful as antidiabetic agents |
| US4812570 | Jul 14, 1987 | Mar 14, 1989 | Takeda Chemical Industries, Ltd. | Method for producing thiazolidinedione derivatives |
| US5554758 | Jun 7, 1995 | Sep 10, 1996 | Takeda Chemical Industries, Ltd. | P-/2-/5-ethyl-2-pyridyl/ethoxy/benzaldehyde |
| US5585495 | Dec 4, 1992 | Dec 17, 1996 | The Upjohn Company | Using a cobalt compound, ligand and reducing agent |
| US5952509 | Jun 23, 1997 | Sep 14, 1999 | Takeda Chemical Industries, Ltd. | Reacting p-hydroxybenzaldehyde with a pyridyl-ethyl sulfonate derivative to form 4-(2-(substituted or nonsubstituted-2-pyridyl)ethoxy) benzaldehyde |
| US6100403 | Apr 12, 1999 | Aug 8, 2000 | Takeda Chemical Industries, Ltd. | Production of benzaldehyde compounds |
| US20020050563 | Sep 14, 2001 | May 2, 2002 | Smithkline Beecham P.L.C. | Process for the preparation of pharmaceutically active thiazolidine or oxazolidine compounds by a yeast reductase |
| US20020106762 | Oct 9, 2001 | Aug 8, 2002 | Smith Kline Beecham Plc | Generating preerential compound for use as prophylactic agent; obtain chemical intermediate, incubate with enzyme, recover prophylactic agent |
| US20060252803 | Jan 26, 2006 | Nov 9, 2006 | Ben-Zion Dolitzky | Hydrogenation of precursors to thiazolidinedione antihyperglycemics |
| US20070078170 | Aug 30, 2004 | Apr 5, 2007 | Khanduri Chandra H | Process for the preparation of pioglitazone |
| US20090118514 | Nov 5, 2008 | May 7, 2009 | Raghupathi Reddy Anumula | Processes for preparing pioglitazone and its pharmaceutically acceptable salts |
| WO2012153312A1 | May 11, 2012 | Nov 15, 2012 | Ranbaxy Laboratories Limited | Process for the purification of pioglitazone |
| EP1734036A1 * | Jun 14, 2005 | Dec 20, 2006 | Well-being Biochemical Corp. | Process for preparation of tamsulosin and its derivatives |
| WO1997031907A1* | Feb 26, 1997 | Sep 4, 1997 | Grady Evan Boswell | Substituted 4-hydroxy-phenylalcanoic acid derivatives with agonist activity to ppar-gamma |
| WO2002088120A1* | Apr 25, 2002 | Jul 11, 2002 | Ales Halama | Method for obtaining pioglitazone as an antidiabetic agent |
| EP0008203A1 * | Aug 3, 1979 | Feb 20, 1980 | Takeda Chemical Industries, Ltd. | Thiazolidine derivatives, preparing same and pharmaceutical compositions comprising same |
| EP0193256A1 * | Jan 15, 1986 | Sep 3, 1986 | Takeda Chemical Industries, Ltd. | Thiazolidinedione derivatives, their production and use |
 NAVEGLITAZAR
2(S)-Methoxy-3-[4-[3-(4-phenoxyphenoxy)propoxy]phenyl]propionic acid
476436-68-7
C25 H26 O6, 422.4703
NAVEGLITAZAR
2(S)-Methoxy-3-[4-[3-(4-phenoxyphenoxy)propoxy]phenyl]propionic acid
476436-68-7
C25 H26 O6, 422.4703
- CCRIS 9448
- LY 519818
- LY 9818
- LY519818
- LY9818
- Naveglitazar
- UNII-Y995M7GM0G
Naveglitazar, a peroxisome proliferator-activated receptor (PPAR) modulator, had been in phase II clinical trials for the once-daily oral treatment of type 2 diabetes, however, no recent development for this indication has been reported. The compound was originally discovered through an ongoing research collaboration between Lilly and Ligand, but, in 2006, Lilly discontinued the development program.
Naveglitazar [LY519818; benzenepropanoic acid, alpha-methoxy-4-[3-(4-phenoxyphenoxy)propoxy], (alpha-S)-] is a nonthiozolidinedione peroxisome proliferator-activated receptor alpha-gamma dual, gamma-dominant agonist that has shown glucose-lowering potential in animal models and in the clinic.
Studies have been conducted to characterize the disposition, metabolism, and excretion of naveglitazar in mice, rats, and monkeys after oral and/or i.v. bolus administration.
………………………………
Martín JA, Brooks DA, Prieto L, González R, Torrado A, Rojo I, López de Uralde B, Lamas C, Ferritto R, Dolores Martín-Ortega M, Agejas J, Parra F, Rizzo JR, Rhodes GA, Robey RL, Alt CA, Wendel SR, Zhang TY, Reifel-Miller A, Montrose-Rafizadeh C, Brozinick JT, Hawkins E, Misener EA, Briere DA, Ardecky R, Fraser JD, Warshawsky AM.
Bioorg Med Chem Lett. 2005 Jan 3;15(1):51-5.
………………………………………..
EXAMPLE 153
′2-Methoxy-3-{3-[3-(4-phenoxy-phenoxy)-propoxy]-phenyl}-propionic acid
 The title compound was prepared from 3-(3-Hydroxy-phenyl)-2-methoxy-propionic acid methyl ester from Example 152, Step D with 4-(3-bromopropoxy)1-phenoxybenzene in a manner analogous as in Example 152, Step E. MS (ES) for C25H26O6[M+NH4]+: 440.2, [M+Na]+: 445.2. 1H-NMR (CDCl3, 200.15 MHz): 7.33-7.17 (m, 3H), 7.07-6.78 (m, 10H), 4.15 (dt, 4H, J=1.9, 6.2), 4.03 (dd, 1H, J=7.3, 4.3), 3.40 (s, 3H), 3.13 (dd, 1H, J=14.2, 4.6), 2.98 (dd, 1H, J=14.0, 7.5), 2.25 (qui, 2H, J=5.9)ppm.
The title compound was prepared from 3-(3-Hydroxy-phenyl)-2-methoxy-propionic acid methyl ester from Example 152, Step D with 4-(3-bromopropoxy)1-phenoxybenzene in a manner analogous as in Example 152, Step E. MS (ES) for C25H26O6[M+NH4]+: 440.2, [M+Na]+: 445.2. 1H-NMR (CDCl3, 200.15 MHz): 7.33-7.17 (m, 3H), 7.07-6.78 (m, 10H), 4.15 (dt, 4H, J=1.9, 6.2), 4.03 (dd, 1H, J=7.3, 4.3), 3.40 (s, 3H), 3.13 (dd, 1H, J=14.2, 4.6), 2.98 (dd, 1H, J=14.0, 7.5), 2.25 (qui, 2H, J=5.9)ppm.
Share this:
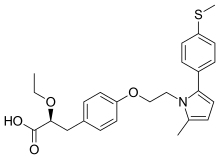 (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
(2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid
(αS)-α-Ethoxy-4-[2-[2-methyl-5-[4-(methylthio)phenyl]-1H-pyrrol-1-yl]ethoxy]benzenepropanoic Acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid
- alpha-ethoxy-4-(2-(2-methyl-5-(4-methylthio)phenyl))-1H-pyrrol-1-yl)ethoxy))benzenepropanoic acid magnesium salt
- saroglitazar
- ZYH1 compound
- E0YMX3S4JD
- cas no 495399-09-2
Saroglitazar, Lipaglyn
- E0YMX3S4JD
- cas no 495399-09-2
| MOLECULAR WEIGHT | 439.56706 g/mol |
|---|---|
| MOLECULAR FORMULA | C25H29NO4S |
 |
| Zydus Cadila chairman and MD Pankaj R. Patel (centre) and deputy managing director Sharvil P. Patel (left) in Mumbai on Wednesday. (PTI)JUNE 5, 2013 |
Calcutta Telegraph
It generally takes around 10-15 years for a drug to be developed from the time of its discovery In the case of Lipaglyn, the molecule was identified in 2001, and Phase III clinical trials was completed around four years ago. While Zydus has not yet…http://www.telegraphindia.com/1130606/jsp/business/story_16976915.jsp

| Saroglitazar, is a drug for the treatment of diabetic dyslipidemia and hypertriglyceridemia with Type 2 diabetes mellitus not controlled by statin therapy. Its trade name is Lipaglyn. It is also a 1,2-Diarylpyrroles derivative, which can be used in the preparation of Nonsteroidal anti-inflammatory drugs (NSAIDs). |
| References: Khanna, I. K., et al.: J. Med. Chem., 40, 1619 (1997) |
 | |
| SYSTEMATIC (IUPAC) NAME | |
|---|---|
| (2S)-2-Ethoxy-3-[4-(2-{2-methyl-5-[4-(methylsulfanyl)phenyl]-1H-pyrrol-1-yl}ethoxy)phenyl]propanoic acid | |
| CLINICAL DATA | |
| TRADE NAMES | Lipaglyn |
| PREGNANCY CAT. |
|
| LEGAL STATUS |
|
| ROUTES | Oral |
| IDENTIFIERS | |
| CAS NUMBER | 495399-09-2 |
| ATC CODE | None |
| PUBCHEM | CID 60151560 |
| CHEMSPIDER | 32079086 |
| CHEMICAL DATA | |
| FORMULA | C25H29NO4S |
| MOL. MASS | 439.56 g/mol |
Mechanism of action
Saroglitazar is novel first in class drug which acts as a dual PPAR agonist at the subtypes α (alpha) and γ (gamma) of theperoxisome proliferator-activated receptor (PPAR). Agonist action at PPARα lowers high blood triglycerides, and agonist action onPPARγ improves insulin resistance and consequently lowers blood sugar.[2]

_MoA.png)
Clinical trials
The approval for saroglitazar was based on the results obtained from clinical studies, which were conducted for more than eight years.
The studies evaluated the efficacy, safety, pharmacokinetics andpharmacodynamics of the drug. Phase I clinical trials on saroglitazar were conducted in 2005. The highest dose of saroglitazar evaluated in a Phase I trial was 128 mg, several times the estimated therapeutic doses (1–4 mg). The pharmacokinetics of saroglitazar support a once daily dosage schedule. No serious adverse events were reported.[3] Phase II studies were completed in 2006.
The Phase III clinical trials were conducted between 2008 and 2011. The first Phase III clinical trials on saroglitazar compared saroglitazar 4 mg dose with pioglitazone 45 mg. The results of the study demonstrated that patients who were administered with saroglitazar 4 mg dose showed reduction in LDL cholesterol and triglycerides, and increase in HDL cholesterol. The study also showed that saroglitazar administered patients showed a reduction in fasting plasma glucose and glycosylated hemoglobin.
Saroglitazar 2 mg and 4 mg significantly reduced (P < 0.001) plasma triglycerides from baseline by 26.4% (absolute change ± SD: −78.2 ± 81.98 mg/dL) and 45% (absolute change ± SD −115.4 ± 68.11 mg/dL), respectively, as compared to pioglitazone -15.5% (absolute change ± SD: −33.3 ± 162.41 mg/dL) at week 24. Saroglitazar 4 mg treatment also demonstrated marked decrease in low-density lipoprotein (5%), very-low-density lipoprotein (45.5%), total cholesterol (7.7%), and apolipoprotein-B (10.9%).[4]
The second Phase III clinical trials on saroglitazar were conducted to evaluate the diabetic dyslipidemic patients insufficiently controlled with statin therapy. The second Phase III study results showed that patients treated with saroglitazar showed pronounced beneficial effect on both the lipid and glycaemic parameters.
At Week 12, saroglitazar 2-mg and 4-mg tablets significantly reduced mean plasma triglyceride levels by -45.5±3.03% and -46.7±3.02% (mean±SE), respectively, and the difference was significant (P<0.001) compared with placebo. Saroglitazar 2 mg demonstrated significant decrease in levels of non-HDL-C, very LDL-C, total cholesterol, and fasting plasma glucose. Additionally, saroglitazar 4 mg also significantly reduced LDL-C and apolipoprotein B levels. Saroglitazar was found to be safe and well tolerated by patients.[5]
Safety
Saroglitazar was found to be safe and well tolerated during the clinical program. In Phase III trials, There was no edema or weight gain reported in any of the study arms. During this study, subjects were monitored for cardiac events, ECG abnormalities, and cardiac function by 2-D ECHO at the start of the study, at the end of 12 weeks, and at 24 weeks after the last dose of the study drug. There were no adverse events reported as far as cardiac safety is concerned.
After 12 weeks of treatment, there were a no significant changes in hemoglobin, liver enzymes (alkaline phosphatase, alanine aminotransferase,aspartate aminotransferase, andγ-glutamyl transferase), renal function (creatinine, enhanced glomerular filtration rate, and blood urea nitrogen), CPK, and high-sensitivity C-reactive protein in the saroglitazar and placebo arms.[6][7]
In Phase I clinical trials saroglitazar was used up to 128 mg and found well tolerated. No serious adverse events were reported. Adverse events were generally mild and moderate in nature and did not show any clinically relevant findings in clinical laboratory investigations, physical examinations, vital signs and electrocardiograms.[8]
…………………………………..
PAPER
A new enantioselective synthesis of (S)-2-ethoxy-3-(4-hydroxyphenyl)propanoic acid esters (EEHP and IEHP), useful pharmaceutical intermediates of PPAR agonists
Tetrahedron Lett 2014, 55(21): 3223
 …………………………………….
WO 2003009841
…………………………………
US 20030236254
………………………….
US 20140099333
………………………………
…………………………………….
WO 2003009841
…………………………………
US 20030236254
………………………….
US 20140099333
………………………………
 (I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
(I)
The compound as claimed in claim 1 wherein R is -SMe and M+ is Mg+2.
The compound of claim 1 is Saroglitazar.
wherein ‘R’ is selected from hydroxy, hydroxyalkyl, acyl, alkoxy, alkylthio, thioalkyl, aryloxy, arylthio and M+ represents suitable metal cations such as Na+, K+, Ca+2, Mg+2 and the like. r .
……………………………
3-Aryl-2-hydroxy propanoic acid derivatives serve as a key intermediate for the synthesis of many pharmaceutically important compounds especially, peroxime proliferator activated receptor (PPAR) agonist.
Optically active 3-aryl-2-alkoxy propanoic acid and its esters, particularly, ethyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (EEHP) and isopropyl (2S)-2-ethoxy-3-(4-hydroxyphenyl)propanoate (IEHP) are versatile chiral pharmacophores present in many pharmaceutically important compounds, especially in peroxisome proliferator activated receptor (PPAR) agonists that have beneficial effects in treating Type 2 diabetes.
Several PPAR agonists, in particular PPAR α/γ dual agonists, commonly termed as glitazars (Ragaglitazar, Tesaglitazar, Navaglitazar etc.), as shown in the figure below were developed by many pharmaceutical companies that have a potential application in the treatment of Type 2 diabetes and dyslipidemia.
However, many of these drugs were discontinued due to their undesirable side effects, but some of them still have great potential [For example, Saraglitazar (LipaglynTM) developed by Zydus Cadila got approval in India for the treatment of diabetic dyslipidemia or hypertriglyceridemia]. Several PPAR α/γ agonists possessing chiral (S)-l moieties are shown below.
 Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.
Tesaglitazar Naveglitazar
In addition, these derivatives find an application in photosensitive materials, sweetening agents, treatment of certain eating disorders etc. Therefore, these compounds have attracted a great deal of attention of synthetic chemists and different methods of preparation of the compound of formula (S)-l have been extensively studied.
Generally, the reported protocols for the synthesis involve chiral pool approaches starting from L-tyrosine and its derivatives (Refer WO 02/24625, US 6559335B2, WO 2003/027084), asymmetric synthesis (Org. Lett. 2005, 7, 1947, US 2007/0149804) and resolution processes using chiral amines or enzymes (WO 2000/026200, WO 2001/11073, Org. Process Res. Dev. 2003, 7, 82, Org. Process Res. Dev. 2004, 8, 838, Tetrahedron Asymmetry 2009, 20, 2594).
Some of these methods have disadvantages such as expensive chiral starting materials and catalysts, low enantioselectivity and overall yields, problems associated with the O-alkylation step which often leads to the loss of optical purity, and many others.
The processes described in WO20026200 (Rao et. al.) uses benzyl bromide for benzylation, which is highly lachrymatory. Again, in the processes described, the debenzylation of the final intermediate was done by using Pd/C under pressure, which escalates the process economics.
WO2003024915 describes a process for the preparation 3-aryl-2-hydroxy propanoic acid derivatives from 3-(4-hydroxyphenyl)-2-oxopropanoic acid.
WO 2003008362 describes 3-Aryl-2-hydroxy propanoic acid derivatives of formula I and the preparation thereof.
 wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1
wherein Rland R2 may be same or different and represent hydrogen or (CI- C6) alkyl.
The process is depicted in Scheme 1 below.
Scheme 1
 In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)
In another process variant as in Scheme 2, WO’362 discloses a process for the preparation of novel 3-aryl-2 -hydroxy propanol and their derivatives of the formula (I)
 wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2
wherein OR and OR together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms, which comprises: i) reducing the compound of formula (III) where R represents hydrogen or alkyl group, R3 represents benzyl to a compound of formula (IV) where R3 represents benzyl, ii) cyclizing the compound of formula (IV) to a compound of formula (V) where ORl and OR2 together form a substituted or unsubstituted 5 membered cyclic structure containing carbon and oxygen atoms and R3 represents benzyl and iii) debenzylating the compound of formula (V) in the presence of metal catalysts to yield pure compound of formula (I).
Scheme 2
 Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).
Both the processes described in WO’362 result in poor overall yield and further fail to describe the preparation of compound of formula V using different alkylating agents. This document exemplifies the compound of formula V with similar ether groups as it fails to teach selective alkylation of formula IV.
WO2005019152 discloses an improved process for the preparation of compound of the general formula (la) and (lb).
 Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3
Wherein, Rl represent H or (C1-C6) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl and the like. R2 represents (Ci-Ce) alkyl group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t- butyl and the like. R3 represents H, protecting groups such as benzyl, substituted benzyl, (C1-C3) alkyl and like.
The compound of general formula (la) is prepared according to the following schemes 3 and 4.
Scheme 3
 Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
Both the processes start with selective O-alkylation or O-aralkylation of L-Tyrosine of formula (2a) using a base, a chelating agent, an alkyl or aralkyl halide in the presence of solvents to obtain the compound of formula (3a), which is diazotized to obtain formula (4a) which upon dialkylation using an excess of alkylating agent and excess base, in presence of suitable solvent to obtain optically pure compound of formula (la). Alternatively, compound of formula (4a) may be selectively esterified to obtain compound of formula (5a), which is subsequently O-alkylated to obtain compound of formula (la) (Scheme 2).
However, the above processes have many disadvantages such as multistep synthesis including protection & deprotection and low overall yield. Further, low temperature diazotization on industrial scale is not viable. Moreover, the starting material is very expensive and hence escalates the process.
In the light of the foregoing, development of a new, alternate enantio-selective synthetic route to these important chiral intermediates, which are simple and can preserve the optical purity at the C-2 carbon of 3-Aryl-2-hydroxy propanoic acid derivatives, is highly desirable. There is a need for an efficient process for synthesis of 3-Aryl-2-hydroxy propanoic acid derivatives of formula (S)-l in high enantiopurity and good overall yield from commercially available starting material.
Tetrahedron Lett 2014, 55(21): 3223
(I)References
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
- “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- 7 “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- 8 “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
CIGLITAZONE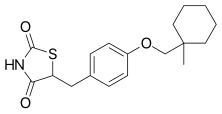
Ciglitazone
U-63287, ADD-3878
- Molecular FormulaC18H23NO3S
- Average mass333.445 Da
- 74772-77-3 [RN]
(±)-5-[4-(1-MethylcCiglitazone (INN) is a thiazolidinedione. Developed by Takeda Pharmaceuticals in the early 1980s, it is considered the prototypical compound for the thiazolidinedione class.[1][2][3][4]
Ciglitazone was never used as a medication, but it sparked interest in the effects of thiazolidinediones. Several analogues were later developed, some of which—such as pioglitazone and troglitazone—made it to the market.[2]
Ciglitazone significantly decreases VEGF production by human granulosa cells in an in vitro study, and may potentially be used in ovarian hyperstimulation syndrome.[5] Ciglitazone is a potent and selective PPARγ ligand. It binds to the PPARγ ligand-binding domain with an EC50 of 3.0 μM. Ciglitazone is active in vivo as an anti-hyperglycemic agent in the ob/ob murine model.[6] Inhibits HUVEC differentiation and angiogenesis and also stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells.[7]
SYN
T. Sohda, K. Mizuno, E. Imamiya, Y. Sugiyama, T. Fujita, and Y. Kawamatsu, Chem. Pharm. Bull., 30, 3580 (1982).

SYN
Ciglitazone (CAS NO.: ), with other name of , 5-((4-((1-methylcyclohexyl)methoxy)phenyl)methyl)-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:

The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol - water.
Syn
Chem Pharm Bull 1982,30(10),3580
The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol - water.
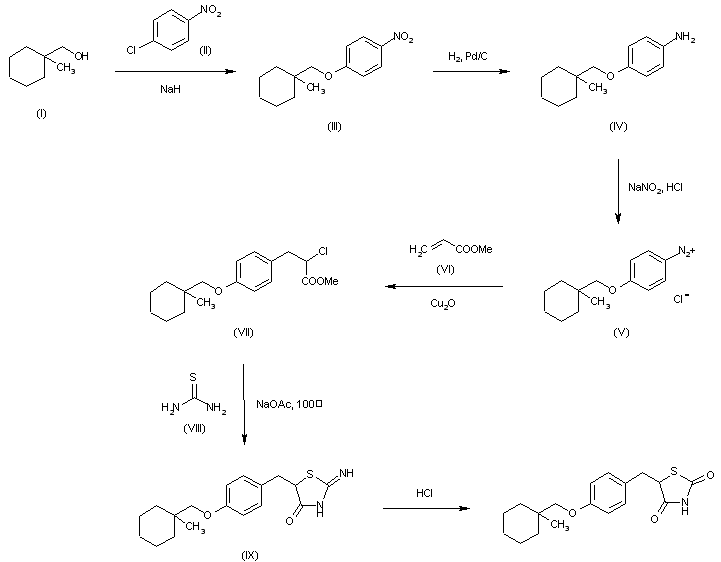
By cyclization of (VIII) with methyl 2-(methanesulfonyloxy)-3-[4-(1-methylcyclohexylmethoxy)phenyl]propionate (X) by means of sodium acetate in hot 2-methoxyethanol, followed by hydrolysis with HCl in ethanol water.

paper
Vijay Kumar Sharma , Anup Barde & Sunita Rattan (2020): A short review on synthetic strategies toward glitazone drugs, Synthetic Communications, DOI: 10.1080/00397911.2020.1821223
Experimental process for synthesis of ciglitazone is fairly robust, albeit pyrophorophic NaH as base is utilized for synthesis of 15.
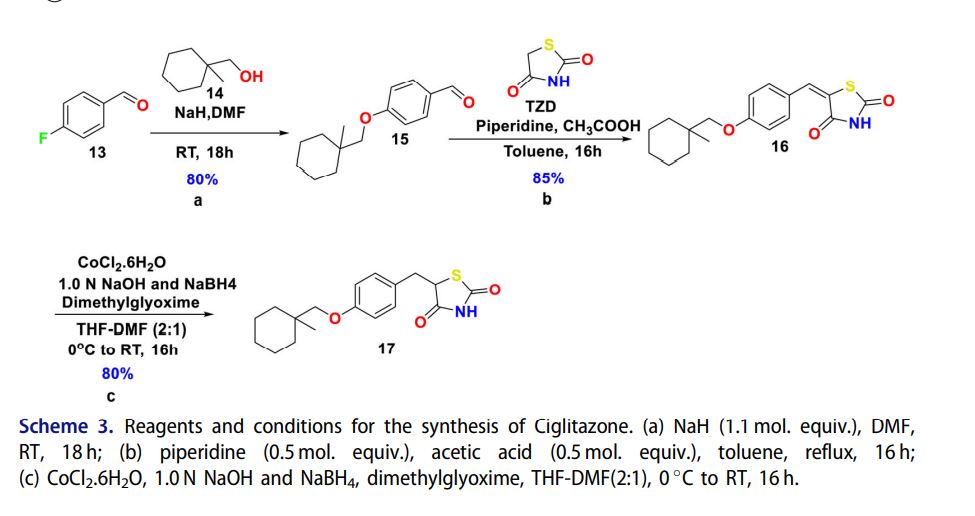
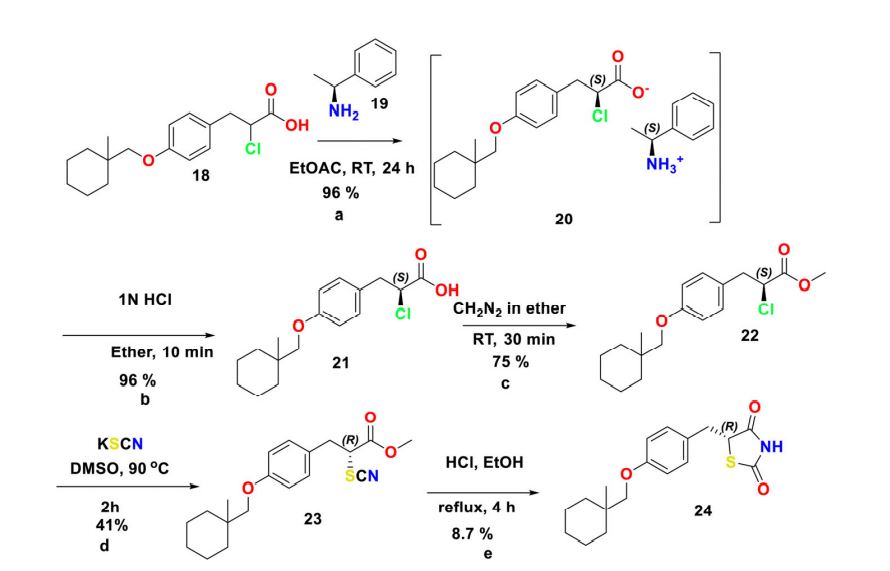
Scheme 4. Reagents and conditions for the preparation of (R)-ciglitazone 24 (a) (S)-1-phenylethan-1- amine 19 (0.9 mol. equiv.), EtOH, RT, 4 h; (b) 1 N HCl (2 vol.), diethyl ether, RT, 10 min; (c) CH2N2 in diethyl ether (ca. 3% w/w), diethyl ether, 0 C-RT, 30 min; (d) KSCN (1.5 mol. equiv.), DMSO, 90 C, 2 h; (e) 2 N HCl (10 vol.), and EtOH, reflux 4 h.
Chiral synthesis Racemic-ciglitazone 17 was resolved with optically active a-methylbenzylamine (PEA) 19 through asymmetric transformation of optical lability at the C-5 position of TZD ring. 2-chloro-3-(4-((1-methylcyclohexyl)methoxy)phenyl)propanoic acid 18 was resolved using (S)-()-1-Phenylethylamine 19 to isolate (S)-2-chloro-3-(4-((1- methylcyclohexyl) methoxy)phenyl)propanoicacid 21. Esterification followed by substitution with KSCN provided methyl (R)-3-(4-((1-methylcyclohexyl)methoxy)phenyl)-2- thiocyanatopropan-oate 23 which was then hydrolyzed to isolate (R)-ciglitazone 24. Similarly, S-isomer was also isolated with (R)-(þ)-1-phenylethylamine (Scheme 4).
[30]Sohda, T.; Mizuno, K.; Kawamatsu, Y. Studies on Antidiabetic Agents. VI. Asymmetric Transformation of (þ/-)-5-[4-(1-Methylcyclohexylmethoxy)Benzyl]-2,4- Thiazolidinedione (Ciglitazone) with Optically Active 1-Phenylethylamines. Chem. Pharm. Bull. 1984, 32, 4460–4465. DOI: 10.1248/cpb.32.4460.
- “Zydus Group launches new diabetic drug”. The Times of India. Jun 6, 2013.
- “Lipaglyn (Saroglitazar) for Treating Hypertriglycerdemia in Type II Diabetes, India”. Drug Development and Technology.
- “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
- “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- “A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI)”. Diabetes Technology and Therapeutics. Feb 2014.
- 7 “A Multicenter, Prospective, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Saroglitazar 2 and 4 mg Compared to Pioglitazone 45 mg in Diabetic Dyslipidemia (PRESS V)”. Journal of Diabetes Science and Technology. Jan 2014.
- 8 “Pharmacokinetics, safety, and tolerability of saroglitazar (ZYH1), a predominantly PPARα agonist with moderate PPARγ agonist activity in healthy human subjects.”. Clinical Drug Investigation. Nov 2013.
Ciglitazone
U-63287, ADD-3878
- Molecular FormulaC18H23NO3S
- Average mass333.445 Da
- 74772-77-3 [RN]
Ciglitazone (INN) is a thiazolidinedione. Developed by Takeda Pharmaceuticals in the early 1980s, it is considered the prototypical compound for the thiazolidinedione class.[1][2][3][4]
Ciglitazone was never used as a medication, but it sparked interest in the effects of thiazolidinediones. Several analogues were later developed, some of which—such as pioglitazone and troglitazone—made it to the market.[2]
Ciglitazone significantly decreases VEGF production by human granulosa cells in an in vitro study, and may potentially be used in ovarian hyperstimulation syndrome.[5] Ciglitazone is a potent and selective PPARγ ligand. It binds to the PPARγ ligand-binding domain with an EC50 of 3.0 μM. Ciglitazone is active in vivo as an anti-hyperglycemic agent in the ob/ob murine model.[6] Inhibits HUVEC differentiation and angiogenesis and also stimulates adipogenesis and decreases osteoblastogenesis in human mesenchymal stem cells.[7]
SYN
T. Sohda, K. Mizuno, E. Imamiya, Y. Sugiyama, T. Fujita, and Y. Kawamatsu, Chem. Pharm. Bull., 30, 3580 (1982).
SYN
Ciglitazone (CAS NO.: ), with other name of , 5-((4-((1-methylcyclohexyl)methoxy)phenyl)methyl)-, (+-)-, could be produced through many synthetic methods.
Following is one of the reaction routes:
The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol - water.
Syn
Chem Pharm Bull 1982,30(10),3580
The reaction of 1-methylcyclohexylmethanol (II) with 4-chloronitrobenzene (III) by means of NaH in hot DMSO gives 4-(1-methylcyclohexylmethoxylnitrobenzene (III), which is reduced with H2 over Pd/C in methanol yielding 4-(1-methylcyclohexylmethoxylaniline (IV). Diazotation of (IV) with NaNO2 and HCl in water affords a solution of the corresponding diazonium chloride (V), which is condensed with methyl acrylate (VI) by means of Cu2O affording methyl 2-chloro-3-[4-(1-methylcyclohexylmethoxyl)phenyl]propionate (VII). The cyclization of (VII) with thiourea (VIII) by means of sodium acetate in hot 2-methoxyethanol gives 2-imino-5-[4-(1-methylcyclohexylmethoxy)benzyl]thiazolidin-4-one (IX), which is finally hydrolyzed with HCl in refluxing 2-methoxyethanol - water.
By cyclization of (VIII) with methyl 2-(methanesulfonyloxy)-3-[4-(1-methylcyclohexylmethoxy)phenyl]propionate (X) by means of sodium acetate in hot 2-methoxyethanol, followed by hydrolysis with HCl in ethanol water.
paper
Vijay Kumar Sharma , Anup Barde & Sunita Rattan (2020): A short review on synthetic strategies toward glitazone drugs, Synthetic Communications, DOI: 10.1080/00397911.2020.1821223
Experimental process for synthesis of ciglitazone is fairly robust, albeit pyrophorophic NaH as base is utilized for synthesis of 15.
Scheme 4. Reagents and conditions for the preparation of (R)-ciglitazone 24 (a) (S)-1-phenylethan-1- amine 19 (0.9 mol. equiv.), EtOH, RT, 4 h; (b) 1 N HCl (2 vol.), diethyl ether, RT, 10 min; (c) CH2N2 in diethyl ether (ca. 3% w/w), diethyl ether, 0 C-RT, 30 min; (d) KSCN (1.5 mol. equiv.), DMSO, 90 C, 2 h; (e) 2 N HCl (10 vol.), and EtOH, reflux 4 h.
Chiral synthesis Racemic-ciglitazone 17 was resolved with optically active a-methylbenzylamine (PEA) 19 through asymmetric transformation of optical lability at the C-5 position of TZD ring. 2-chloro-3-(4-((1-methylcyclohexyl)methoxy)phenyl)propanoic acid 18 was resolved using (S)-()-1-Phenylethylamine 19 to isolate (S)-2-chloro-3-(4-((1- methylcyclohexyl) methoxy)phenyl)propanoicacid 21. Esterification followed by substitution with KSCN provided methyl (R)-3-(4-((1-methylcyclohexyl)methoxy)phenyl)-2- thiocyanatopropan-oate 23 which was then hydrolyzed to isolate (R)-ciglitazone 24. Similarly, S-isomer was also isolated with (R)-(þ)-1-phenylethylamine (Scheme 4).
[30]Sohda, T.; Mizuno, K.; Kawamatsu, Y. Studies on Antidiabetic Agents. VI. Asymmetric Transformation of (þ/-)-5-[4-(1-Methylcyclohexylmethoxy)Benzyl]-2,4- Thiazolidinedione (Ciglitazone) with Optically Active 1-Phenylethylamines. Chem. Pharm. Bull. 1984, 32, 4460–4465. DOI: 10.1248/cpb.32.4460.
References
- ^ Pershadsingh HA, Szollosi J, Benson S, Hyun WC, Feuerstein BG, Kurtz TW (June 1993). "Effects of ciglitazone on blood pressure and intracellular calcium metabolism". Hypertension. 21 (6 Pt 2): 1020–3. doi:10.1161/01.hyp.21.6.1020. PMID 8505086.
- ^ Jump up to:a b Hulin B, McCarthy PA, Gibbs EM (1996). "The glitazone family of antidiabetic agents". Current Pharmaceutical Design. 2: 85–102.
- ^ Imoto H, Imamiya E, Momose Y, Sugiyama Y, Kimura H, Sohda T (October 2002). "Studies on non-thiazolidinedione antidiabetic agents. 1. Discovery of novel oxyiminoacetic acid derivatives". Chem. Pharm. Bull. 50 (10): 1349–57. doi:10.1248/cpb.50.1349. PMID 12372861.
- ^ Sohda T, Kawamatsu Y, Fujita T, Meguro K, Ikeda H (November 2002). "[Discovery and development of a new insulin sensitizing agent, pioglitazone]". Yakugaku Zasshi (in Japanese). 122 (11): 909–18. doi:10.1248/yakushi.122.909. PMID 12440149.
- ^ Shah DK, Menon KM, Cabrera LM, Vahratian A, Kavoussi SK, Lebovic DI (April 2010). "Thiazolidinediones decrease vascular endothelial growth factor (VEGF) production by human luteinized granulosa cells in vitro". Fertil. Steril. 93 (6): 2042–7. doi:10.1016/j.fertnstert.2009.02.059. PMC 2847675. PMID 19342033.
- ^ Willson, T.M.; Cobb, J.E.; Cowan, D.J.; et al. (1996). "The structure-activity relationship between peroxisome proliferator-activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones". J Med Chem. 39 (3): 665–668. doi:10.1021/jm950395a. PMID 8576907.
- ^ Xin, X.; et al. (1999). "Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo;". J. Biol. Chem. 274 (13): 9116–21. doi:10.1074/jbc.274.13.9116. PMID 10085162.
Ciglitazone Clinical data ATC code - none
Identifiers CAS Number PubChem CID IUPHAR/BPS DrugBank ChemSpider UNII KEGG ChEMBL CompTox Dashboard (EPA) ECHA InfoCard 100.220.474 
Chemical and physical data Formula C18H23NO3S Molar mass 333.45 g·mol−1 3D model (JSmol)
/////////ciglitazone, U 63287, ADD 3878, DIABETES
by WORLD DRUG TRACKER
DR ANTHONY
- ^ Pershadsingh HA, Szollosi J, Benson S, Hyun WC, Feuerstein BG, Kurtz TW (June 1993). "Effects of ciglitazone on blood pressure and intracellular calcium metabolism". Hypertension. 21 (6 Pt 2): 1020–3. doi:10.1161/01.hyp.21.6.1020. PMID 8505086.
- ^ Jump up to:a b Hulin B, McCarthy PA, Gibbs EM (1996). "The glitazone family of antidiabetic agents". Current Pharmaceutical Design. 2: 85–102.
- ^ Imoto H, Imamiya E, Momose Y, Sugiyama Y, Kimura H, Sohda T (October 2002). "Studies on non-thiazolidinedione antidiabetic agents. 1. Discovery of novel oxyiminoacetic acid derivatives". Chem. Pharm. Bull. 50 (10): 1349–57. doi:10.1248/cpb.50.1349. PMID 12372861.
- ^ Sohda T, Kawamatsu Y, Fujita T, Meguro K, Ikeda H (November 2002). "[Discovery and development of a new insulin sensitizing agent, pioglitazone]". Yakugaku Zasshi (in Japanese). 122 (11): 909–18. doi:10.1248/yakushi.122.909. PMID 12440149.
- ^ Shah DK, Menon KM, Cabrera LM, Vahratian A, Kavoussi SK, Lebovic DI (April 2010). "Thiazolidinediones decrease vascular endothelial growth factor (VEGF) production by human luteinized granulosa cells in vitro". Fertil. Steril. 93 (6): 2042–7. doi:10.1016/j.fertnstert.2009.02.059. PMC 2847675. PMID 19342033.
- ^ Willson, T.M.; Cobb, J.E.; Cowan, D.J.; et al. (1996). "The structure-activity relationship between peroxisome proliferator-activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones". J Med Chem. 39 (3): 665–668. doi:10.1021/jm950395a. PMID 8576907.
- ^ Xin, X.; et al. (1999). "Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo;". J. Biol. Chem. 274 (13): 9116–21. doi:10.1074/jbc.274.13.9116. PMID 10085162.
 | |
| Clinical data | |
|---|---|
| ATC code |
|
| Identifiers | |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.220.474 |
| Chemical and physical data | |
| Formula | C18H23NO3S |
| Molar mass | 333.45 g·mol−1 |
| 3D model (JSmol) | |
/////////ciglitazone, U 63287, ADD 3878, DIABETES
DR ANTHONY
No comments:
Post a Comment